
 |
 |
 |
 |
 |
 |
 |
 |
ExonMine is a Perl program used to mine mRNA and EST datasets for alternative splicing. The webserver’s underlying MySQL databases provide information on alternative splicing in the following 13 model organisms:
Human - Homo sapiens
Chimp - Pan troglodytes
Rhesus - Macaca mulatta
Mouse - Mus musculus
Rat - Rattus norvegicus
Cow - Bos taurus
Dog - Canis lupus familiaris
Zebrafish - Danio rerio
Chicken - Gallus gallus
Frog - Xenopus tropicalis
Fly - Drosophila melanogaster
Nematode worm - Caenorhabditis elegans
Sea squirt - Ciona intestinalis
The output data formats are intended to aid in laboratory validation and microarrays.
ExonMine data is searchable using gene symbol or any GenBank accession via a friendly interface containing five useful laboratory day-to-day tools: In silico PCR; primer cross-amplification check; search for isoform-specific sequences for primer design; search with transcript sequence; decomposition of input transcript sequence into ExonMine exons. It also provides downloadable tab delimited tables with sequence data for microarray prode design linked to alternative splicing events. (TOP)
BLAT alignments of mRNAs and spliced ESTs to a genome assembly are used to obtain information of exon/intron boundaries. Tables of such alignments are available from the UCSC Genome Browser. The genomic region of the gene is determined using RefSeq BLAT alignments associated with a given EntrezGeneID and extended to any mRNAs or ESTs that splice to exons in the RefSeq alignments.
BLAT alignments consist of a series of aligned sequence blocks separated by gaps of genomic sequence One would normally expect the aligned blocks to correspond to exons and the gaps in the alignment to correspond to introns; however, this is not always the case and the program proceeds with a filtering out of data which we do not consider of sufficiently good quality to permit extraction of information on alternative splicing: two adjacent blocks separated by gaps smaller than 30nt are not considered to be reliable introns and are joined into a single block. These small gaps can be due to short repeats of variable length in the genomic sequence and the cDNA or to short stretches of the cDNA which the BLAT program did not succeed in aligning due to single nucleotide polymorphisms, these are refered to as ‘query inserts’. A record of the position of all these ‘filled in’ regions is kept and is available for each gene.
Filtering of captured mRNA and EST BLAT alignments allows only: and minimum intervening sequence size of 30 nucleotides; splice site consensus sequences GT...AG, GC...AG, or AT...AC; and minimum exon of 9 nucleotides. Where these requirements are not met an alignment is cut into fragments at the non-consensus junction or on either side of the small exon, and only those fragments with more than one exon are kept; intervening sequences sorter than 30 nucleotides are ‘filled in’ . Each alignment is corrected to the nearest known spliced junction or, for the terminal regions, to the longest corresponding block, according to the set of pre-established rules.The resulting blocks are considered to correspond to exons and the intervening gaps to introns and each sequence is assigned a number and type. The minimal set of longest splicing patterns covering all the collected spliced junctions, is retrieved. A reference sequence is used to apply a numbering system on the exons based on the numbering of exons in the first known splicing pattern detected for a particular gene. The representative set is then fed into a directed graph object used to retrieve a description of all alternative splicing events. The description of alternative splicing events is provided by comparing pairs of isofoms. Information on orthologs, constitutive exons, polyadenylaton sites and all open reading frames is given. Every mRNA and EST collected for each gene is associated with the sequential numbering of exons; using simple pattern matching the user can determine all the accessions, sex, development or tissue which contain a particular exon, junction or splicing pattern. (TOP)
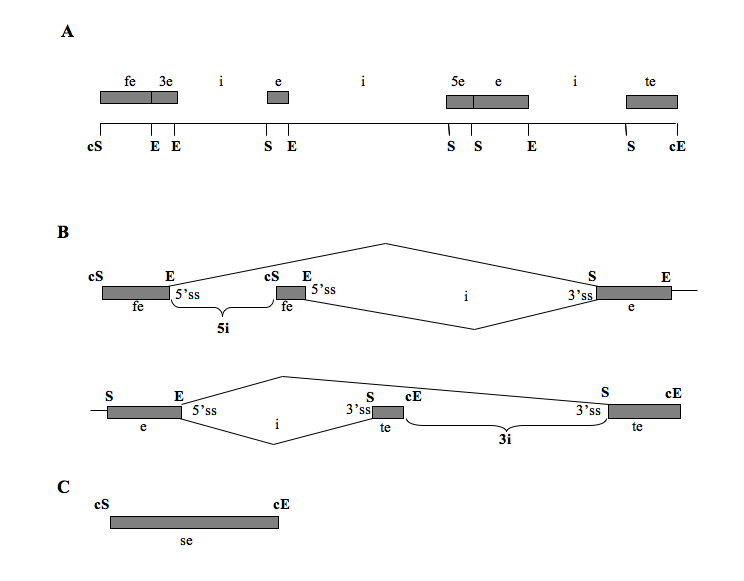
The nomenclature use to describe the organisation of the gene using the known starts and ends of all superimposed exons is shown in Figure 1.
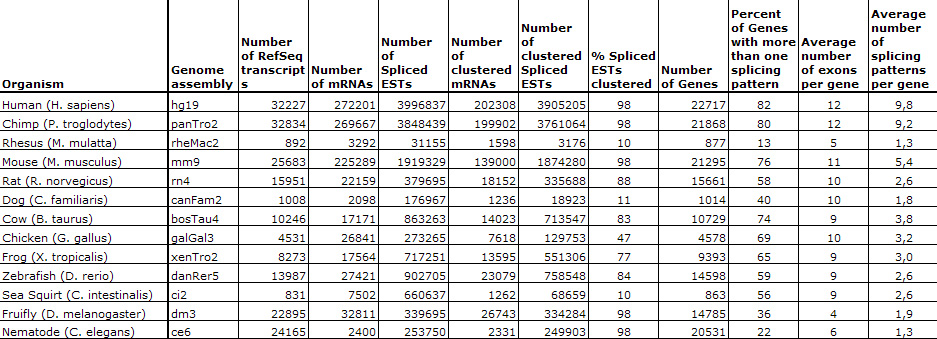
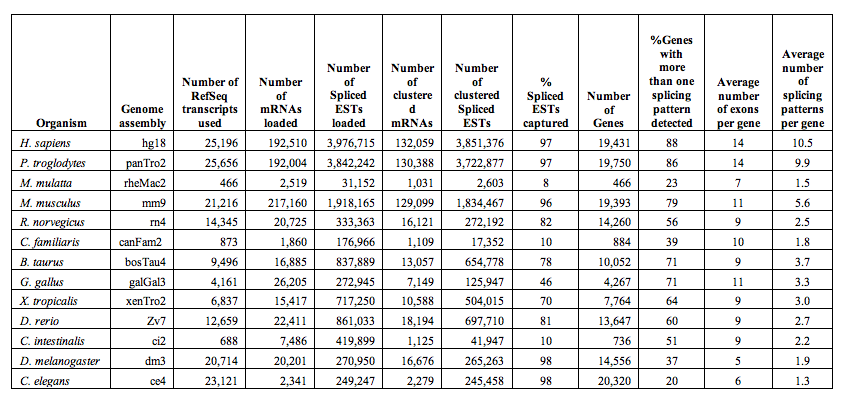
Statistics for the January 2010 update
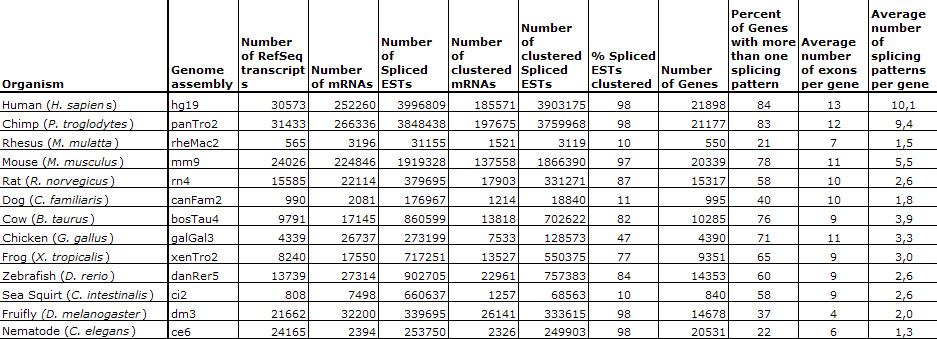
Statistics for the September 2009 update
Statistics for the May 2009 update
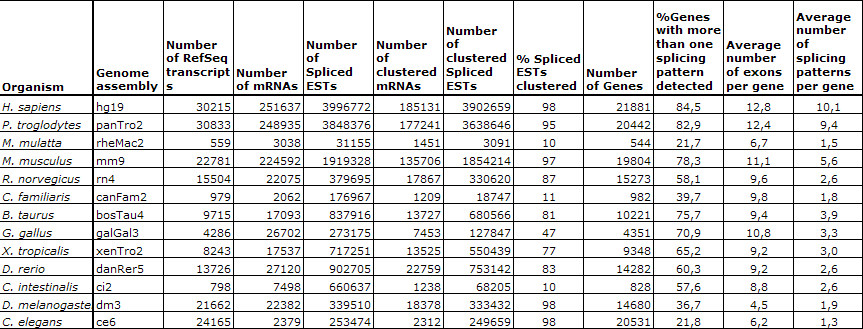
Statistics for the January 2009 update
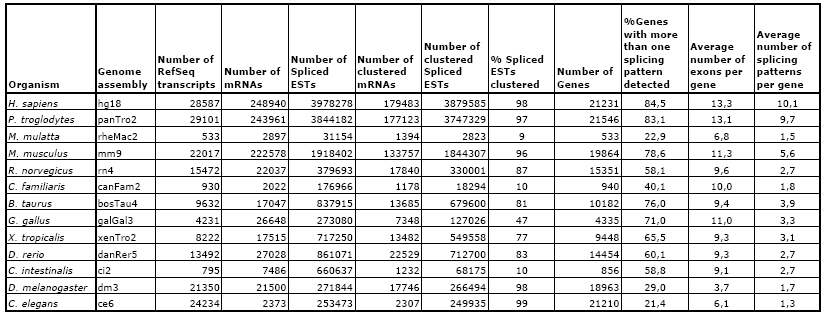
Statistics for the August 2008 update
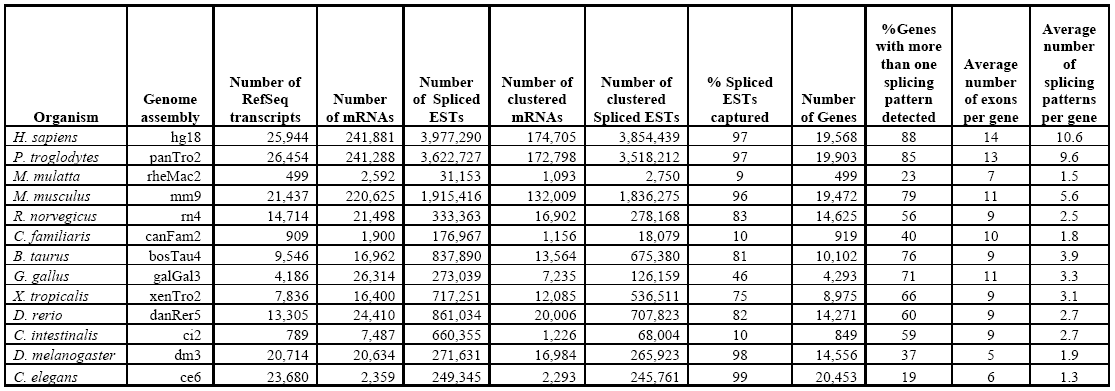
Statistics for the April 2008 update
Basic search
To query ExonMine, the user selects an organism and searches for a Gene using the Gene Symbol ID or a GenBank Accession. Useful information guiding the user is given when the mouse passes over information (i) points on the screen. ExonMine output provides information in both graphical and downloadable formats and it is organized into five main fields. The first display gives the results of the initial search and indicates the organism, genomic location and Entrez Gene link for enties of the queried gene. The user chooses an entry requests the entry to be processed.
The graphical display is divided in two main parts one with gene organization and the other with the transcripts: the gene graphical display shows all exons, introns and their respective types and length; the splicing patterns are shown in the transcript graphical display. Constitutive, Novel, First, Terminal, Upstream and Downstream Exons are colour coded. The mRNA and/or EST accessions corresponding to each complete splicing pattern are also indicated. For each gene we also provide the orthologue prediction in other organisms and a link that will perform automatically the basic search for the orthologue in the chosen organism. All information is presented in new windows to facilitate comparison between searches. Finally, several downloadable options are available for sequences in fasta format for isoforms, exons and intron; or tables with all information relatively to exons, introns, isoforms (including presence of poly-A signals), translation (start and stop codons, and reading frames), expression specificity (information related with biological source of mRNA/EST). Searches for tissue expression for any exon, junction or splicing pattern can be performed.
The user can also download tables containing junction sequences (50 nt centered on the junction) involved in alternative splicing events, for microarray probe design.
Additional Tools
The web interface provides five additional tools: an “In silico PCR” tool that checks for primers cross-amplification; a search for isoform-specific sequences for primer design; a search for matches given an input transcript sequence; and the possibility of decomposing an input transcript sequence into ExonMine exons. The tools were developed using R language and packages RMySQL and seqinr [Team et al 2007]. For the “In silico PCR” tool the user enters the Gene Symbol ID and primer sequences and ExonMine graphically represents the primers, identifies the splicing patterns and the fragment length that would be amplified by the submitted primers (Figure 4E). The user can also verify if the primers cross-amplify transcripts from other genes using the second tool.
ExonMine also allows the search for isoform-specific sequences for primer design given a GeneSymboID and an isoform number. In this case the partial sequence is returned with the specific exon-junction in uppercase and the corresponding exons identified.
Finally, the user can input a transcript sequence and search for matching isoforms in ExonMine or decompose the submitted transcript sequence into fragments (exons, introns, or unknown fragments). (TOP)